INTRODUCTION
There have been cases reporting either duplication of 7q36 to the terminus or deletion of 9p24; however none with both simultaneously. Here, we report a patient with simultaneous de novo 7q36.1-q36.3 duplication and 9p24.3 deletion. A 6-year-old boy was referred for the evaluation of developmental delay. He had microcephaly and mild dysmorphic features, such as a long face and small nose. Chromosome and array comparative genomic hybridization analyses revealed 46,XY,dup(7)(q36.1-q36.3) and del(9)(p24.3). The sizes of the duplication and deletion were 9.9 Mb and 1.9 Mb, respectively. The duplication of chromosome 7 contained 68 known genes, 3 of which are related to entries in the Developmental Disorders Genotype-to-Phenotype (DDG2P) database. The deletion of chromosome 9 contained 6 known genes, 2 of which are in the DDG2P database. This study investigated the genotype and phenotype in this patient, and reviewed the relevant literatures for possible clinical presentations in these variations.
CASE REPORT
A 6-year-old boy was transferred to our institution from a local hospital for evaluation for developmental delays in speech and language. He could communicate using about 10 words, but could not form sentences. He could not read or write, and was only able to draw circles. The patient showed some growth delay as well; his weight was 17.8 kg (<3rd percentile), height 115.8 cm (10th–15th percentile), and head circumference 48.5 cm (<3rd percentile). He was born preterm at 35 weeks of gestational age; however there was no other prepartum or postpartum event. His birth weight was 2,400 g (<3rd percentile). When he was 2 years old, operations for both inferior oblique muscle over-action with exotropia were performed. Until the age of 3 years, he had been hospitalized several times due to recurrent episodes of pneumonia. He started to walk with hand support at 23 months old, and independent outdoor gait was possible when he was 36 months old. He received speech and sensory integration therapy at the private center, and was enrolled in kindergarten for disabled children 3 years ago. His parents were healthy, and there was no remarkable finding in his pedigree. His siblings (one sister and one brother) showed normal development.
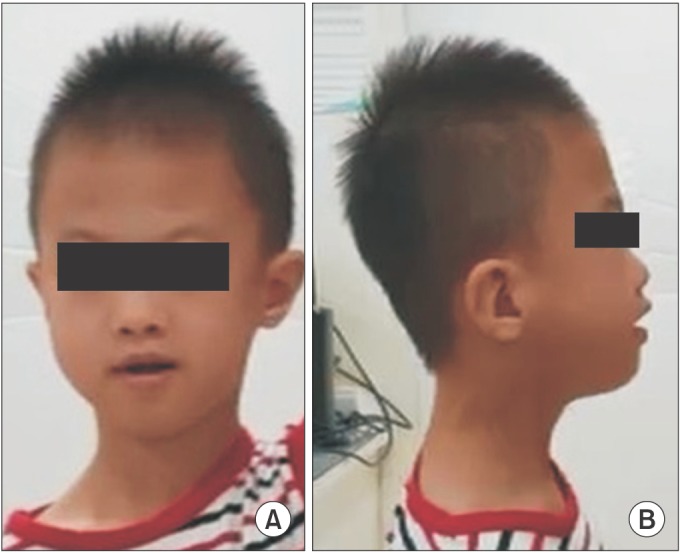
When he visited the clinic, he could walk upstairs, but he couldn't tiptoe or jump. In pediatric evaluation of disability inventory, the normative standard scores of self-care, mobility, and social function were 15.0, 24.4, and 14.5, respectively. In addition, he received a score of 38.65 in the social quotient. All of them were below 2 standard deviations. In the Bayley Scale of Infant and Toddler Development 3rd edition, the age equivalents for gross motor, fine motor, and cognition were 31, 27, and 27 months, respectively. The age equivalents for receptive and expressive language were 24 and 18 months, respectively, according to the sequenced language scale for infants. The results of the receptive and vocabulary test were below the 10th percentile, and the score for the Korean oral syntax expression comprehension test was below the 1st percentile. He could imitate only 1 syllable. The patient had microcephaly and mild facial dysmorphic features, specifically a long face with high forehead, as well as a small nose and small ears (Fig. 1). On physical examination, deep tendon reflexes were normal, and no upper motor neuron signs were detected. Brain magnetic resonance imaging (MRI) indicated no definite abnormalities.
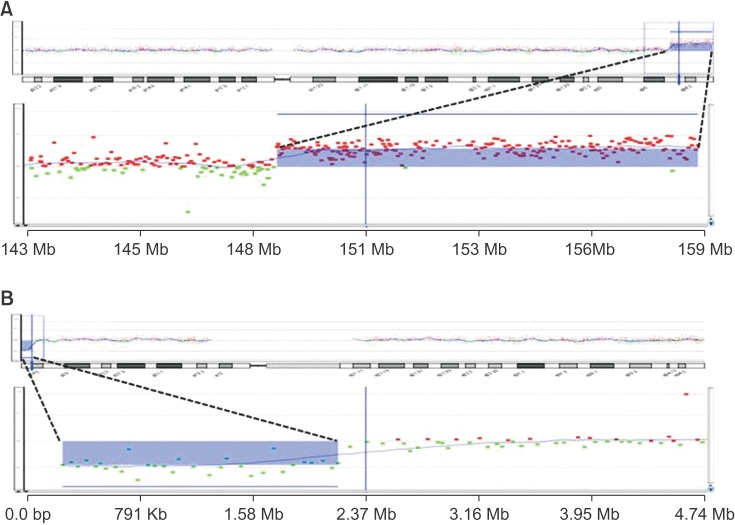
Laboratory studies did not reveal any specific findings, such as Wilson's disease, autoimmune disease, endocrine disease, or metabolic disease. Chromosome analysis and array comparative genomic hybridization (CGH) analysis showed 46,XY,dup(7)(q36.1-q36.3) and del(9) (p24.3) (Fig. 2). The duplication on chromosome 7, spanning 149,128,443–159,088,636 bp, was estimated to be 9.9 Mb in size, containing 68 known genes, while the deletion of chromosome 9, spanning 271,257–2,183,334 bp, was calculated to be 1.9 Mb in size and included 6 known genes (Table 1). The genetic information is described on the following websites: Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway), Online Mendelian Inheritance in Man (OMIM, http://www.ncbi.nlm.nih.gov/omim) [1], DECIPHER (http://decipher.sanger.ac.uk) [2], and Ensembl genome browser (http://www.ensembl.org/homo_sapiens/location). Chromosome analysis and array CGH analysis of both parents were performed, and no specific findings were observed.
DISCUSSION
Global developmental delay and intellectual disability are relatively common pediatric conditions, affecting 3% of the general population. Furthermore, up to 40% of such developmental disability cases are caused by genetic factors. The American and European guidelines on this group state that genetic testing is recommended as a standardized diagnostic practice [3]. In addition, Miller et al. [4] recommend chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental disability. Establishing the cause of a child's disability can predict prognosis and other possible complications. Furthermore, it can assist in counselling parents and providing choices for therapeutic and educational interventions, as well as rehabilitation. Therefore, genetic study, including array CGH should be recommended for unexplained developmental delay or intellectual disability such as the case presented in this study.
Phenotypes of a few patients with the 7q36.1-q36.3 duplication or 9p24.3 deletion have been reported. However, to our knowledge, this is the first report of a patient harboring both copy number variations. Therefore, the genotype–phenotype correlation has not been clearly defined. The findings in our patient were compared to those of previously reported cases.
The 9p deletion syndrome was first described in 1973. It is characterized by multiorgan syndrome, which includes dysmorphic facial features (e.g., trigonocephaly, midface hypoplasia, and long philtrum), hyperlaxity with frequent abdominal hernia, abnormalities of the extremities, and variable degrees of cognitive delay [5]. The deletion breakpoint—heterogeneous and variable in size—occurs from 9p22 to 9p24. In the present patient, the size of the deletion in chromosome 9 was 1.9 Mb. The 6 known genes in the 1.9-Mb deletion of chromosome 9 were described in Table 1. Some of the patient's characteristics were associated with 9p deletion syndrome, e.g., midface hypoplasia, long philtrum, and intellectual disability.
Deletion of the short arm of chromosome 9 is also associated with sexual development disorders, and the region was recently further narrowed down to 9p24.3-pter, containing the doublesex- and mab-3-related transcription factor (DMRT) domain. DMRT genes are the strongest candidates for the gonadal dysgenesis phenotype, however the underlying molecular mechanism remains unclear [6]. A patient with complete 46,XY gonadal dysgenesis and motor developmental retardation underwent a gonadectomy at 3.2 years of age [7], and histological analysis disclosed dysgenetic gonads with gonadoblastoma. The present patient shows normal external genitalia, and endocrine laboratory studies, including sex hormone analysis, showed findings within the normal range for his age.
Another gene in the deletion site of chromosome 9, dedicator of cytokinesis 8 (DOCK8, OMIM 611432), is expressed in the immune system, and mutation in this gene results in immune-related disorders [1]. Engelhardt et al. [8] reported the clinical phenotype of 64 patients with DOCK8 deficiency, and uncovered an association with frequent infection and high mortality at a young age. It is therefore noteworthy that the present patient had been hospitalized several times until 3 years due to recurrent episodes of pneumonia. Immunological evaluation was performed at our clinic, but the results were normal.
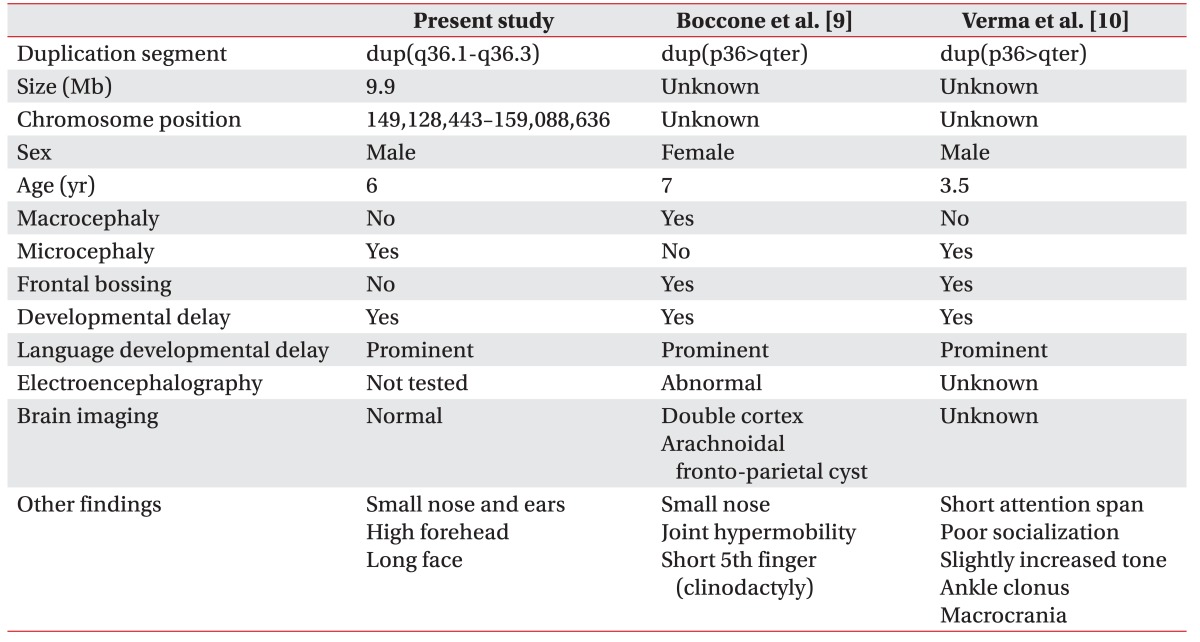
In the present patient, the duplication of chromosome 7 was estimated to be 9.9 Mb in size, and it contained 68 genes, including 3 genes from the DDG2P, namely motor neuron and pancreas homeobox 1 (MNX1), sonic hedgehog (SHH), and WD repeat-containing protein 60 (WDR60). Two patients with duplication from 7q36 to the chromosome terminus have been reported [9,10], and both children showed developmental delay, significantly delayed speech and language, as well as frontal bossing (Table 2). Our patient had severe language and speech delay, however brain MRI showed no significant findings.
MNX1, SHH, and WDR60 are in the DDG2P database because variants in these genes are associated with specific developmental phenotypes. MNX1 (OMIM 142994) encodes a transcription factor containing a homeobox domain. Loss-of-function mutations in MNX1 are associated with Currarino syndrome, which is characterized by anorectal, sacral, and presacral anomalies [1]. SHH (OMIM 600725) encodes a protein implicated in the patterning of the ventral neural tube, anterior-posterior limb axis, and ventral somites in the early embryo. SHH mutations have been found to underlie the following comorbidities: holoprosencephaly-3, single median maxillary central incisor, schizencephaly, and microphthalmia with coloboma-5 [1]. WDR60 (OMIM 615462) encodes a member of the WD repeat protein family, which is involved in a variety of cellular processes such as cell cycle progression, signal transduction, and apoptosis. Mutations in this gene have been found to be related to short-rib polydactyly and Jeune syndrome [1].
The work presented here reports the first case of concurrent de novo duplication of 7q36.1-q36.3 and deletion of 9p24.3. Although some characteristics were similar to those of previously reported cases, no specific genotype-phenotype correlations were established. Further cumulative data based on the molecular approach are warranted to understand the role and influence of the genes in the 7q36.1-q36.3 duplication and 9p24.3 deletion regions. Additionally, if a national registration system for sharing genetic information had existed in Korea, multi-center studies with larger subjects would have been easily possible, and this concept could prove beneficial for future studies.