INTRODUCTION
Stroke is one of the leading causes of death and disability worldwide. As the survival rate of stroke increases, the importance of treatment for disability is emerging [1]. One of the causes of disability in stroke survivors is upper limb spasticity, which can cause significant functional impairment, pain, and reduced quality of life [2].
Treatment options for upper limb spasticity include stretching, electrical stimulation, nerve block, and surgery. Medications for oral spasticity are frequently used, although the responses vary among patients and systemic side effects may occur. Local treatment, namely, botulinum toxin injection, which blocks release of acetylcholine at the neuromuscular junction, is used widely to manage upper limb spasticity after stroke [3].
NABOTA (Daewoong botulinum toxin type A; Daewoong Pharmaceutical, Seoul, Korea) is a novel botulinum toxin type A formulation that is derived from wild-type Clostridium botulinum Hall A. NABOTA is produced by strictly controlled anaerobic fermentation and high-efficiency purification through size-exclusion high-performance liquid chromatography with a single 900 kDa peak (>98%) [4].
The efficacy and safety of NABOTA were investigated in a previous in vivo study [5]. Moreover, a phase III double-blind randomized controlled trial that included 197 patients with stroke found that the efficacy and safety of NABOTA were non-inferior to that of Botox (Onabotulinum toxin A; Allergan Inc., Irvine, CA, USA) [6]. After these studies, NABOTA was approved on December 7, 2015 by the Ministry of Food and Drug Safety in South Korea for use in the indication of upper limb spasticity in stroke patients aged ≥18 years.
With the introduction of newer botulinum toxin type A products, post-marketing evaluation of the safety and efficacy of these products is essential to develop standardized injection protocols and to be able to recognize potential side effects. Therefore, in this study, we investigated the effectiveness of NABOTA in relief of upper limb spasticity and any possible adverse effects associated with the injection.
MATERIALS AND METHODS
Study design and participants
This phase IV clinical study has a prospective, multicenter, open-label design and was performed between June 2019 and December 2019 at seven university hospitals (Seoul National University Boramae Medical Center, Seoul National University Hospital, Severance Hospital, Gangnam Severance Hospital, Dongguk University Ilsan Hospital, National Health Insurance Service Ilsan Hospital, and Presbyterian (Jesus) Medical Center) in South Korea.
The study was approved by the Institutional Review Board of each institution (Seoul National University Boramae Medical Center IRB no. 16-2013-82, Seoul National University Hospital IRB no. 1905-007-1032, Severance Hospital IRB no. 4-2019-0478, Gangnam Severance Hospital IRB no. 3-2016-0261, Dongguk University Ilsan Hospital IRB no. 2019-05-002-019, National Health Insurance Service Ilsan Hospital IRB no. 2019-04-028, and Presbyterian (Jesus) Medical Center IRB no. 2019-04-012) and the Ministry of Food and Drug Safety and performed in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. All subjects provided written informed consent after receiving a detailed explanation about the research.
Study participants were required to be aged ≥18 years, to be at least 6 weeks from onset of stroke, and to have significant symptoms of upper limb spasticity that required treatment. However, the Modified Ashworth Scale (MAS) and Disability Assessment Scale (DAS) score were not considered as inclusion criteria because we aimed to confirm the efficacy and safety of NABOTA in a general clinical practice environment.
Patients who had hypersensitivity to any ingredient in NABOTA, those with a diagnosis of neuromuscular junction or motor neuron disease, and those who were pregnant or breastfeeding or had a possibility of pregnancy were excluded.
Study protocol and treatment
The subjects visited the clinic on the day of the injection (baseline) and at 4, 8, and 12 weeks post-injection. NABOTA was injected once on the first day under ultrasound or electromyographic guidance. Each vial of NABOTA (100 units) was diluted with 2–5 mL of 0.9% sodium chloride. The following five muscles were injected: biceps brachii, flexor digitorum profundus, flexor digitorum sublimis, flexor carpi ulnaris, and flexor carpi radialis. A total of 100–200 units were injected at up to 4 sites in biceps brachii, 15–60 units at 1–2 sites in flexor carpi radialis, and 15–50 units at 1–2 sites in the other muscles (Table 1). The amount of NABOTA injected was decided according to the degree of spasticity and study protocol. The maximum amount of NABOTA injected was 360 units in each patient.
During the study period, patients were required not to take antibiotics (especially aminoglycosides) or anti-cholinergics that could interfere with the efficacy of NA-BOTA. However, muscle relaxants and benzodiazepines were allowed at a fixed dose for >4 weeks before study enrollment. Patients taking muscle relaxants or benzodiazepines were permitted to continue on the same dose throughout the study period.
Since treatments including physical therapy, occupational therapy, splinting, stretching, and positioning can influence the spasticity, patients were required to maintain the same treatment for >4 weeks before enrollment and during the study period.
Outcome measures
Outcome measures were the MAS, DAS, and Caregiver Burden Scale (CBS) scores, which were evaluated at each visit, and the Global Assessment Scale (GAS) score, which was assessed at the final visit (week 12). Spasticity of the wrist, elbow, and finger flexor muscles was measured as the MAS score by physicians [7]. Subjects were placed in a supine position in a comfortable environment for measurement of spasticity. To reduce variability, all physicians used the same methods and the same physician assessed the MAS score for each patient from baseline to final visit [8]. The therapeutic response in terms of spasticity was defined as an improvement in MAS score in comparison with baseline. Functional impairment in hand hygiene, dressing, limb positioning, and pain were rated using the DAS scale (0, no disability; 1, mild disability; 2, moderate disability; 3, severe disability). In this study, one domain was selected per patient by patient/caregiver interview. The CBS scored the extent of the physical burden on the caregiver on a 5-point Likert scale (0, no difficulty; 4, cannot perform) for four items (cleaning the palms, cutting fingernails, dressing, and cleaning the armpits). The GAS score was assessed once at week 12 by both the physician and subject (or caregiver) to measure the perceived benefit of treatment. The MAS, DAS, and CBS scores have been shown to be valid and reliable, and the GAS have been used in several other studies [6,8–10].
The primary study outcome was the change in MAS score for the wrist flexors between baseline and 4 weeks post-injection. The secondary outcomes were as follows: change in MAS score for the wrist flexors at 8 and 12 weeks post-injection from baseline; changes in MAS scores for the elbow and finger flexors at 4, 8, and 12 weeks post-injection from baseline; therapeutic response rates in the wrist, elbow, and finger flexors at 4, 8, and 12 weeks post-injection; change in DAS score at 4, 8, and 12 weeks post-injection from baseline; change in CBS score at 4, 8, and 12 weeks post-injection; and GAS score at 12 weeks post-injection.
Safety evaluation
All subjects were evaluated for any symptoms or unexpected events at each visit. All adverse events were recorded during the study period and coded using the Medical Dictionary for Regulatory Activities version 22 (MedDRA, https://www.meddra.org/). Any abnormality in laboratory results, vital signs, or physical examination was also recorded. Physicians analyzed the relationship between each event and the drug, as well as its severity and seriousness. Adverse events were classified into general events and adverse drug reactions. Events that were fatal or life-threatening, caused permanent disability, or required long-term hospital care were defined as serious adverse events [11].
Statistical analysis
The efficacy data were analyzed in the full analysis set (FAS) and the per-protocol set (PPS). The FAS included all subjects who were eligible and could visit to receive the initial assessment. The PPS was defined as those who were assessed for primary and secondary outcomes without any missing data and followed the study protocol without any violations. The safety of NABOTA was analyzed in the safety set, which was defined as those who had received NABOTA injection in this clinical trial.
Values for the primary and secondary outcomes are presented as the mean and standard deviation. To calculate the mean MAS score, grade 1+ was regarded as 1.5. The Friedman test was used to analyze the changes in outcome variables (MAS grade of upper limb, DAS, and CBS) from baseline to week 4, 8, and 12, given that they showed a non-normal distribution. The Wilcoxon signed-rank test with Bonferroni adjustment was used as a post-hoc test to the Friedman test. We also analyzed the correlation between the GAS scores and the differences in outcome variables between baseline and week 12 using Spearman correlation analysis. All statistical analyses were performed using SPSS version 26 software (IBM Corp., Armonk, NY, USA). The level of significance was set at a p-value of <0.05.
RESULTS
Baseline characteristics
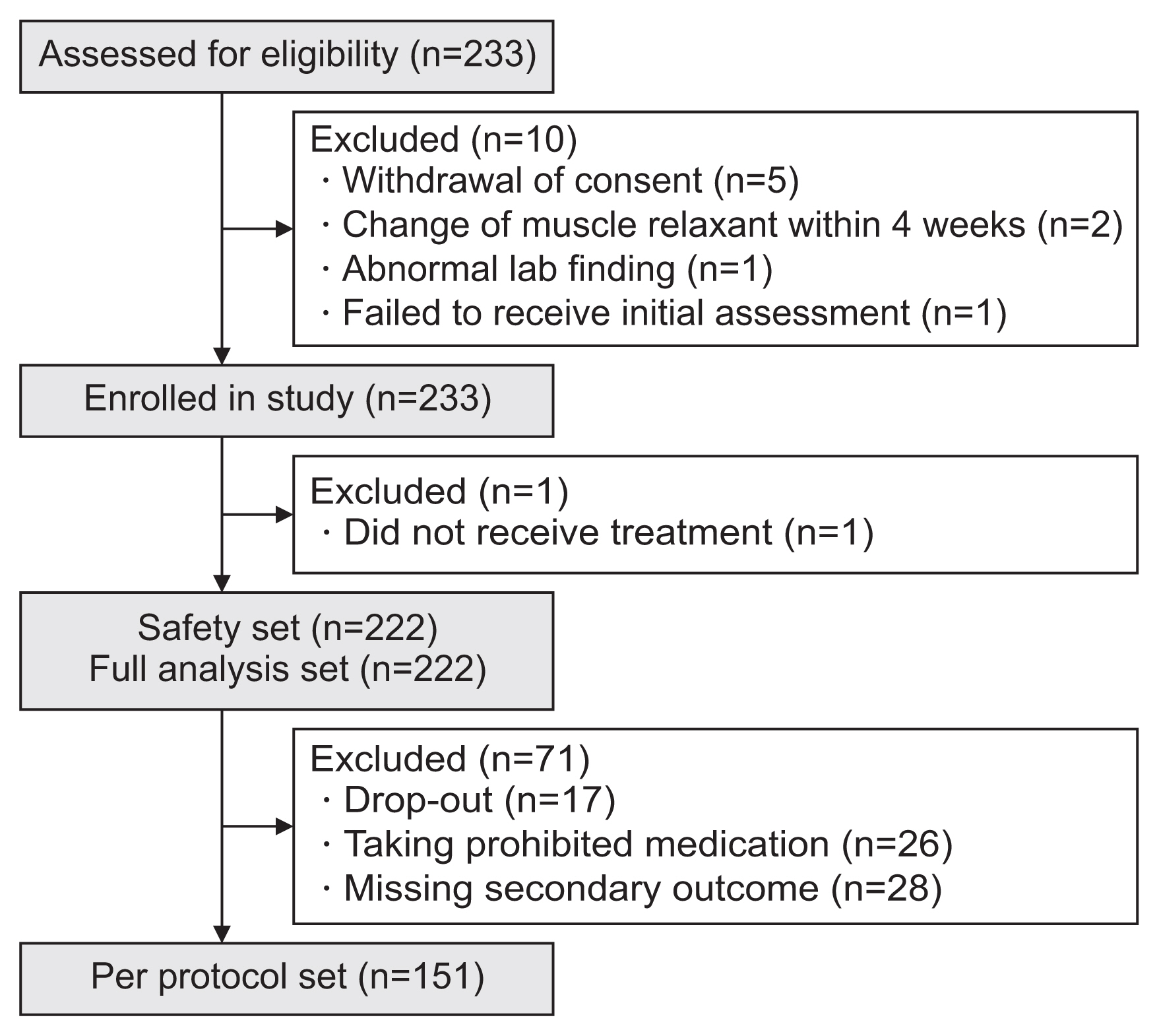
Ten of the 233 patients did not meet the study eligibility criteria and another patient did not receive the study drug, which left 222 patients for inclusion in the FAS and for the safety analysis. Seventy-one further patients were excluded—dropouts (n=17), taking prohibited medication (n=26), a missing secondary outcome (n=28), leaving 151 patients for inclusion in the PPS (Fig. 1).
The 222 subjects included in the FAS had a mean age of 59.5±12.0 years and 75.7% were male. More than half of the patients had hypertension, and 41.0% were receiving concomitant medications during the study period. Among those receiving comcomitant medications, 81 patients took muscle relaxant—baclofen (n=49), dantrolene (n=50), and eperisone (n=6)—and 20 patients took benzodiazepines. Most (99.1%) of the patients had not been treated with botulinum toxin before the study (Table 2).
Primary outcome
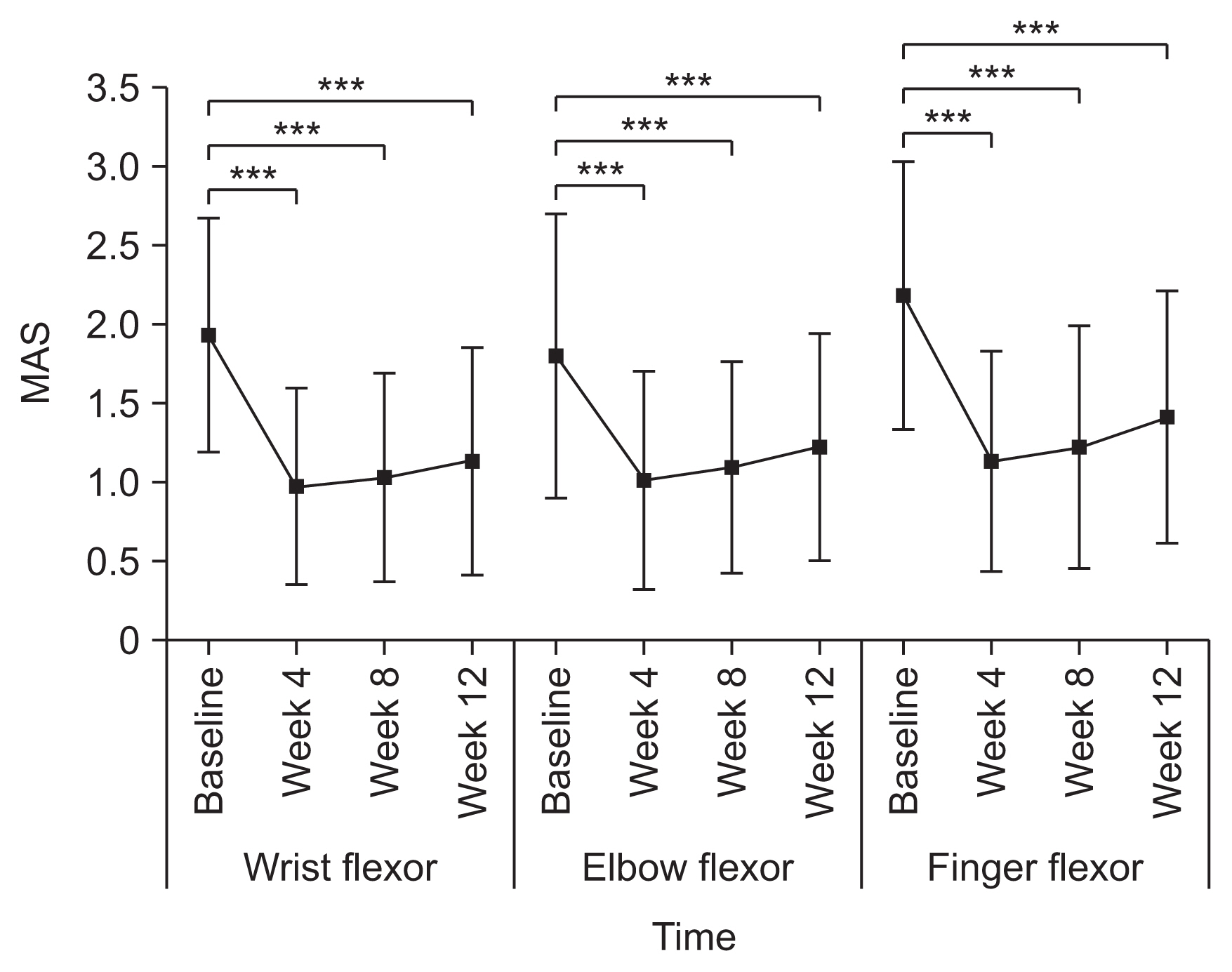
In the FAS, Friedman test showed a significant interaction of time on the wrist flexor MAS score (p<0.001). Post-hoc analysis showed a statistically significant change in the MAS score for the wrist flexors from 1.93±0.74 at baseline to 0.97±0.62 at week 4 (−0.97±0.66, p<0.001) (Fig. 2, Table 3), in the FAS. The PPS analysis also showed a statistically significant interaction of time on the MAS score for the wrist flexors (p<0.001). The change in the wrist flexor MAS score from baseline to week 4 was statistically significant in the PPS analysis (−0.97±0.70, p<0.001) (Supplementary Table S1).
Secondary outcomes
Friedman test for MAS scores for elbow and finger flexors of FAS showed a significant interaction of time on each MAS score (elbow flexor, p<0.001; finger flexor, p<0.001). Post hoc analysis of the FAS showed a significant decrease in the MAS scores for the elbow and finger flexors at week 4 compared with baseline (elbow flexors, −0.79±0.68, p<0.001; finger flexors, −1.06±0.72, p<0.001) (Fig. 2, Table 3). The MAS scores for all injected muscles showed the greatest decrease at the week 4 visit and then a gradual increase until week 12. In the PPS analysis, Friedman test for MAS scores also showed significant interaction of time on each MAS scores (wrist flexor, p<0.001; elbow flexor, p<0.001; finger flexor, p<0.001). Analysis of the PPS showed similarly significant changes and trends in the MAS score for all injected muscles (Supplementary Table S1).
The FAS analysis also showed that the therapeutic response rate was highest at 4 weeks post-injection (wrist flexors, 85.7%; elbow flexors, 77.0%; finger flexors, 88.5%) and decreased gradually thereafter in all muscle groups (Table 4). Similar changes in the therapeutic response rate were observed in analysis of the PPS (Supplementary Table S2).
On the DAS, upper limb position (n=104) was the domain in which improvement was most wanted by patients/caregivers, followed by the hand hygiene (n=60), dressing (n=32), and pain (n=17) domains (Supplementary Table S3). In the FAS, Friedman test for DAS showed significant interaction of time on all DAS domains (Hand hygiene, p<0.001; Dressing, p<0.001; Limb position, p<0.001; Pain, p<0.001) (Supplementary Table S3). Post hoc analysis of the FAS showed that the scores for all domains on the DAS related to principal therapeutic targets were significantly decreased at week 4 compared with baseline (Fig. 3, Supplementary Table S3). Improvements in the DAS scores were maintained (limb position, hand hygiene, and dressing domains) or showed a gradual increase (pain domain) during the follow-up period (Fig. 3, Supplementary Table S3). Like the FAS analysis, the PPS analysis revealed a significant interaction of time on all DAS domains (Hand hygiene, p<0.001; Dressing, p<0.001; Limb position, p<0.001; Pain, p<0.001) and improvement in the DAS score after injection of NABOTA, although there was no statistically significant change between baseline and week 4 for the pain domain (Supplementary Table S3).
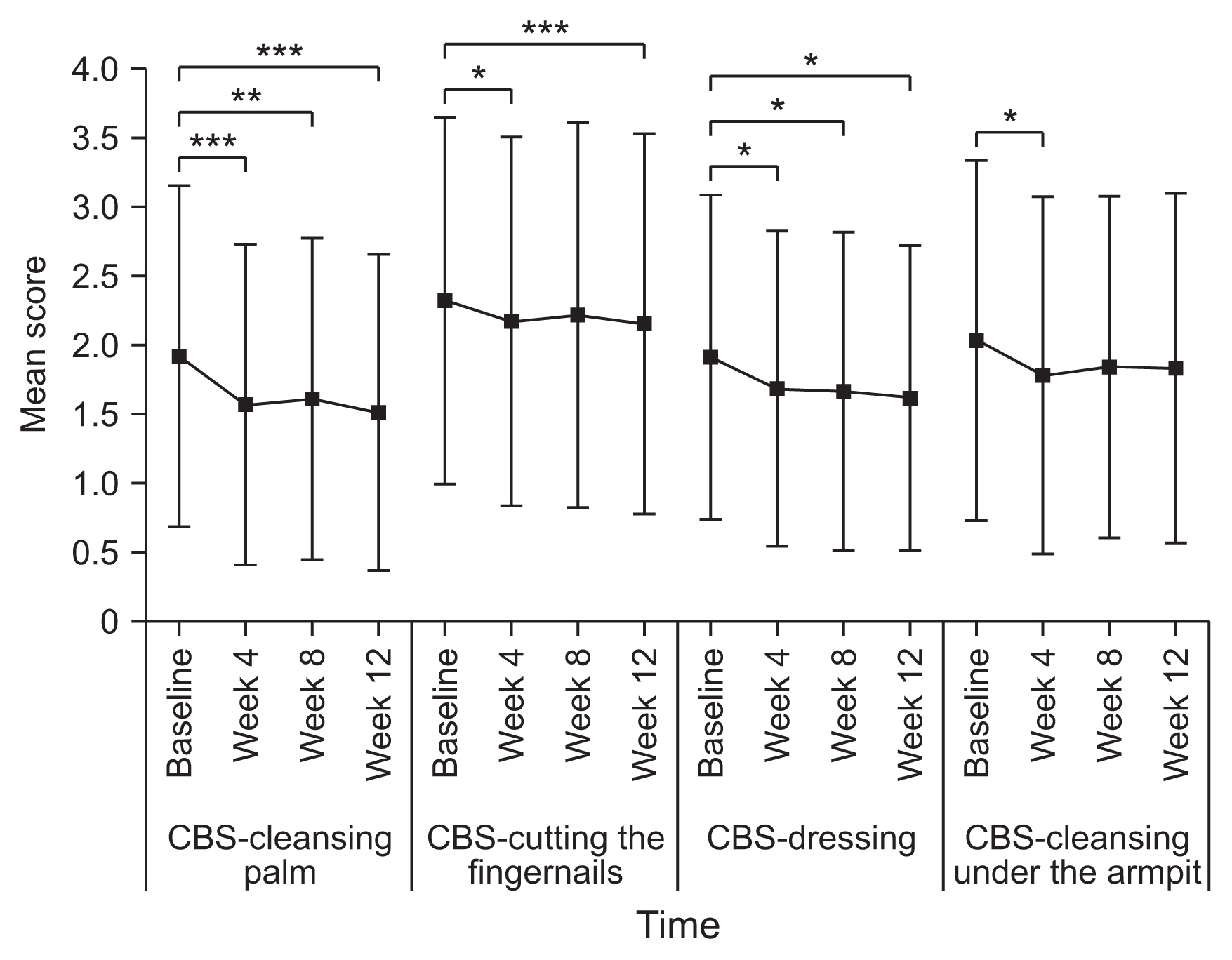
The Friedman test for FAS analysis showed that there was significant interaction of time on CBS scores for all domains (Cleaning the palm, p=0.002; Cutting fingernails, p=0.003; Dressing, p=0.03; Cleaning the armpit, p=0.03). In the post-hoc analysis, CBS scores were significantly decreased in all domains at week 4 (Supplementary Table S4, Fig. 4). However, the changes from baseline were not statistically significant at week 8 for cutting the fingernails domain and at week 8 and 12 for cleaning the armpit domain. The results for the PPS were similar to those for the FAS, although the statistical significances were generally less significant (Supplementary Table S4, Fig. 4).
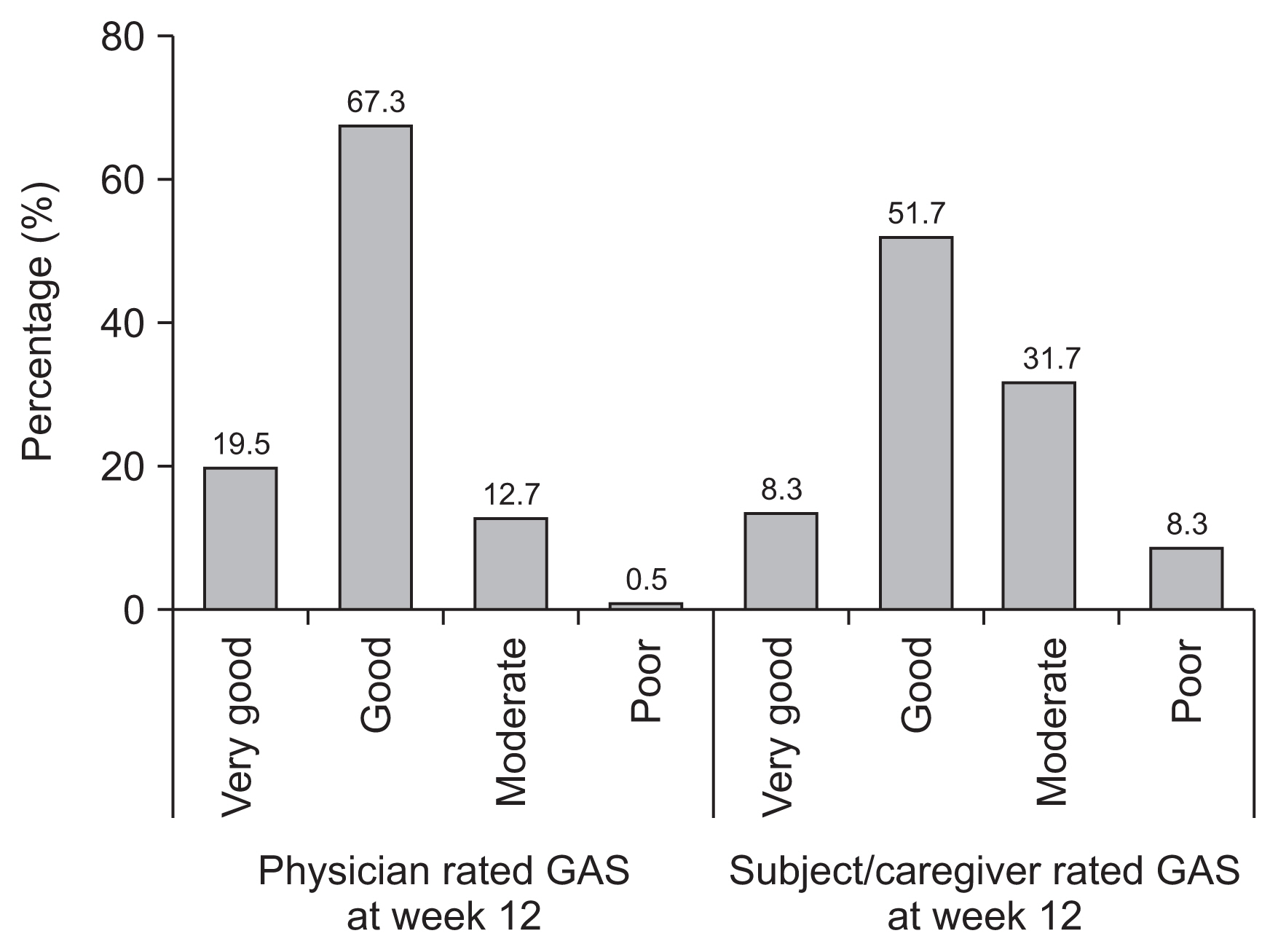
At week 12, the physicians rated the GAS score as very good or good in 86.8% of subjects; however, only 60.0% of the subjects or caregivers rated the GAS score as very good or good at 12 weeks (Supplementary Table S5, Fig. 5).
Safety assessment
The incidence of adverse events was 14.4% (32/222), with 43 events in total. The most frequently reported events were seizure (3 in 3 subjects) and abnormal perception (3 in 2 subjects). Headache, nasopharyngitis, urinary tract infection, fever, and cough were reported in 2 cases from 2 subjects, for each. No adverse drug reactions were reported. Five serious adverse events were reported in 5 subjects. One of the serious adverse events that resulted on dropping out of the trial was pneumonia. Other major adverse events included seizure (2 patients), subdural hemorrhage (1 patient), and fever (1 patient) (Table 5). There were no clinically significant changes in vital signs or findings on physical examination during the study period.
DISCUSSION
This multicenter phase IV study is the first to evaluate the efficacy and safety of NABOTA injections in stroke patients with upper limb spasticity and found significant improvement in their spasticity and related functional disability and a reduction of caregiver burden for up to 12 weeks. The safety assessment showed an incidence of 14.4% for any adverse event. The efficacy of NABOTA was validated for a larger population (n=222) in the present study than in the previous phase III trial (n=92) [6].
There was a significant decrease in the MAS score for all muscles injected at week 4 compared with baseline (wrist flexors, −0.97±0.66; elbow flexors, −0.79±0.68; finger flexors, −1.06±0.72). However, these changes were smaller than the changes in the phase III trial (wrist flexors, −1.44; elbow flexors, −0.73; finger flexors, −1.27). This smaller change in the MAS score likely reflects the broader inclusion criteria and milder spasticity in the present study; the phase III trial included only patients with a wrist flexor MAS score ≥2 while our study included patients with MAS score less than 2 [6]. The effects of treatment decreased gradually after week 4 but persisted until week 12, as in previous studies [6,12,13]. The duration of effect of botulinum toxin A injection in this study is consistent with a previous report suggesting a duration of effect of 3–4 months [14].
In this study, we also found a significant functional improvement after NABOTA injection in terms of the DAS domains of limb positioning, hand hygiene, and dressing. Our results are similar to those in previous studies, including the phase III trial [6,12]. However, the improvement in upper limb function should be interpreted cautiously, given that the DAS domains mainly evaluate simple and basic tasks. Previous studies also demonstrated significant functional improvement after botulinum toxin injection but were limited to passive functions and failed to show gain in active upper limb functions [15,16].
We found that the DAS score in the pain domain was significantly improved after NABOTA injection (week 4, −1.12±0.78; week 8, −1.56±0.51; week 12, −1.53±0.83). Our previous studies could not show the effect of NABOTA on pain because the pain domain was assessed in only a small number of patients (one in the phase III trial of NA-BOTA and four in the Neuronox study) [6,12]. Numerous studies have found that botulinum toxin injection has a significant effect on spasticity-related pain [15–18]. Therefore, like other types of botulinum toxin, NABOTA can be an effective option for management of this type of pain.
The CBS score improved significantly regardless of time and domain, but the changes were small, ranging from approximately 0.3 to 0.5, like in the phase III trial [6]. Similar to the phase III trial, there was still a gap in the GAS score between the physicians and subjects/caregivers. The GAS was rated as very good or good by more than 80% of physicians but by only 60% of patients/caregivers [6]. The discrepancy in GAS scores between physicians and patient/caregivers might reflect the fact that physicians focus more on spasticity while patients/caregivers may put more weight on caregiver burden and more complex upper limb function. We tested this possibility by correlation analysis (Supplementary Table S6), which showed significant correlations between the physician-rated GAS score and the change in MAS score for the upper limb (wrist flexors, p=0.004; finger flexors, p<0.001) and between subject/caregiver-rated GAS and the change in CBS score (cleaning the armpit domain, p=0.001). The change in DAS scores was significantly correlated with both the physician-rated GAS (hand hygiene domain of the DAS, p=0.002) and subject/caregiver-rated GAS (limb position domain of the DAS, p<0.001). Therefore, the caregiver burden and disability should be carefully considered in the management of spasticity, in addition to spasticity itself.
The rate of adverse events in our present study was 14.4%, which is lower than the rate of 19.6% in the previous phase III trial [6]. Moreover, there were no adverse drug reactions in this study whereas three adverse drug reactions were reported in the earlier trial [6]. The Neuronox study reported a higher rate of adverse events (41.5%), with the most common adverse events being nasopharyngitis, extremity pain, and cough [12]. A study of Meditoxin reported an adverse event rate of 35.7%, which mostly consisted of diarrhea, vomiting, edema, nasopharyngitis, and pain [13]. Therefore, we can assume that NABOTA has a safety profile similar to or better than that of other botulinum toxins, although further studies are needed.
This study had several limitations. First, like the previous phase III trial [6], efficacy and safety was evaluated for only 12 weeks. We may have been able to obtain more concrete evidence if the study had been performed with a more extended follow-up period. A study of another botulinum toxin type A preparation showed that the effect was reduced to that at baseline by 10–16 weeks post-injection [19]. Further studies with longer follow-up periods may provide more information on the duration of efficacy of NABOTA. Second, there may have been considerable variability in spasticity because it was evaluated manually. However, the physicians used a uniform measurement protocol and the same physician measured spasticity in each patient from baseline to the final visit in an effort to minimize any variability. Third, we did not include a placebo group, which could have provided more robust evidence of the drug’s efficacy and determined more clearly the possible association between adverse events and injection of NABOTA.
In conclusion, this study found that NABOTA, a novel botulinum toxin type A, has considerable efficacy in the management of upper limb spasticity in stroke patients as well as a good duration of effect. It also demonstrated the safety of NABOTA after its launch on the market. NA-BOTA may be an effective and safe option for management of upper limb spasticity after stroke.