An Infantile Case of Sandhoff Disease Presenting With Swallowing Difficulty
Article information

Abstract
Infants with Sandhoff disease typically appear normal until 3–6 months of age. As the disease progresses, they present with symptoms such as loss of motor skills, exaggerated startle response to loud noise, seizures, visual loss, and paralysis. We encountered a rare case of a 22-month-old girl with Sandhoff disease characterized by progressive motor weakness and dysphagia, who initially showed signs of aspiration at 20 months of age. The major problems related to dysphagia were oromotor dysfunction and abnormal feeding posture. Within 3 months of identification of difficulty in swallowing, the patient showed a significant decrease in food intake, with rapid deterioration of nutritional status. We report our case with a review of the literature.
INTRODUCTION
Sandhoff disease is a rare autosomal recessive lipid storage disorder, with an estimated incidence of approximately 1 in 384,000 live births, and it has three clinical subtypes; infantile, juvenile and adult forms [1]. The infantile form is characterized by progressive neurologic impairment, hypertonia, cherry red spots, and bilateral thalamic involvement [2]. Sandhoff disease is diagnosed by hexosaminidase assay in fibroblasts, white blood cells, or serum using artificial substrates, which shows markedly increased hexosaminidase A and decreased total hexosaminidase [3].
We encountered a rare case of infantile Sandhoff disease exhibiting spasticity and swallowing difficulty. According to a review of literatures, there are only a few reports of an infantile case of Sandhoff disease presenting with dysphagia. But there are no reports about its clinical manifestations.
CASE REPORT
A 22-month-old girl diagnosed with Sandhoff disease was referred to our hospital with chief complaints of spasticity and swallowing difficulty as well as difficulty with head control. She was born at full term through normal vaginal delivery. She was able to stand while holding on to something at 8 months of age, which suggested normal development. However, developmental arrest occurred at 13 months of age. Spasticity and genu recurvatum were observed while standing and holding on to something. The patient showed a progressive motor regression pattern, and at 18 months of age, she could barely maintain a propped sitting position. At 20 months of age, she showed symptoms of aspiration while swallowing and prolonged feeding time.
Brain magnetic resonance imaging scans obtained at 15 months of age did not show delayed myelination or abnormal enhancement (Fig. 1). Developmental regression was similar to that in GM2 gangliosidosis, such as Tay-Sachs disease and Sandhoff disease. The patient underwent hexosaminidase assay in white blood cells, which showed increased hexosaminidase A (%) and decreased total hexosaminidase. The total hexosaminidase A & B and hexosaminidase A levels were 321.2 nmol/hr/mg (reference range, 620–1,000 nmol/hr/mg) and 74.3% (reference range, 55%–72%), respectively. Based on these findings, the patient was diagnosed with Sandhoff disease. However, the patient did not show cherry red spot on fundoscopy.
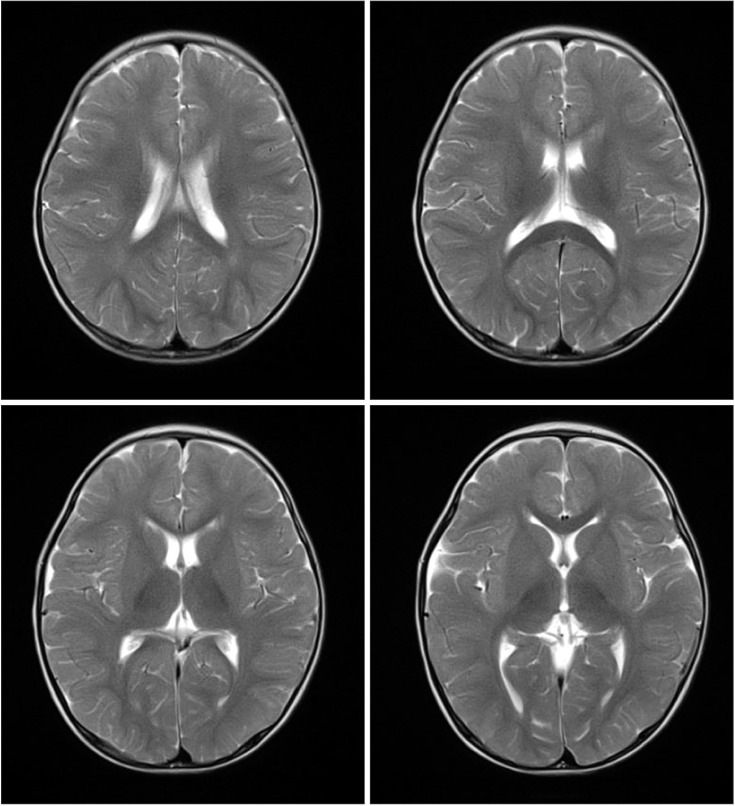
Brain magnetic resonance imaging scans at 15 months of age. On axial T2-weighted images, there are no high-intensity signals in bilateral thalami and basal ganglia.
At the time of admission, physical examination revealed grade 3 and 2 muscle strength in the upper and lower extremities, respectively. Neurological examination showed sustained ankle clonus and increased deep tendon reflexes in all extremities as well as hypertonicity and decerebrate posture manifested by an exaggerated extensor posture of all extremities, while crying and/or when she was irritable (Fig. 2). In terms of spasticity, her four extremities scored grade 2 based on the modified Ashworth scale (MAS).

Physical examination findings. The patient had difficulty in raising her head, turning over, and crawling.
Overall, the patient had developmental regression. Especially, she showed a delay in development by 1 month in gross-motor domain, by 3 months in language and fine motor domain, and by 2 months in personal-social interaction domain in the Denver Developmental Screening Test II.
Dietary assessment revealed that the patient irregularly ingested a soft and blended diet of 70–130 kcal, five times a day, and the mean eating duration was approximately 1 hour. She had difficulty in eating and she showed signs of aspiration (e.g., coughing, gagging). Moreover, she had a poor feeding posture with inadequate trunk and head support.
Oral motor function assessment showed that the patient had increased tone and decreased movement of the tongue and hypersensitivity in the oral and perioral areas. The patient underwent videofluoroscopic swallowing study (VFSS), and was given barium-containing free water, yogurt (thick liquid), and rice porridge (soft). In the oral phase, motor control, including lip sealing, tongue control, and mastication, was inadequate with delayed oral transit time (1.3 seconds) and premature bolus loss [4]. In the pharyngeal phase, a delayed swallow reflex was observed without aspiration or penetration during or after swallowing (Fig. 3).
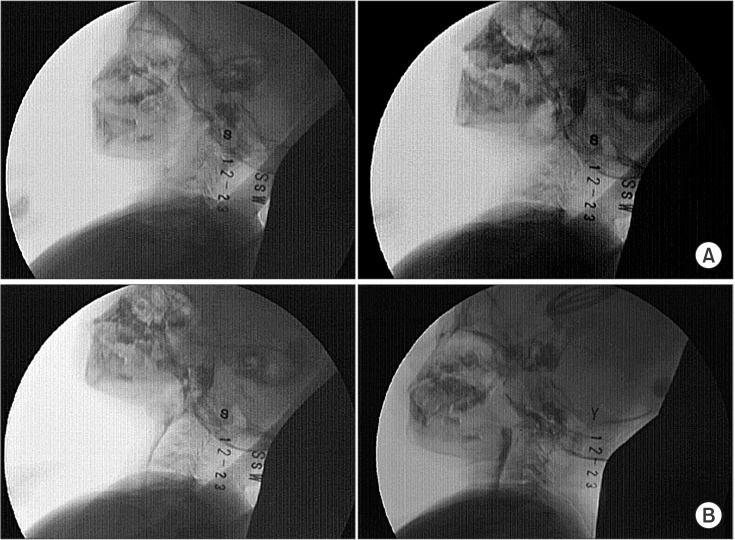
Videofluoroscopic swallowing study findings. (A) No lip closure and lingual elevation in the oral phase. (B) Swallowing without aspiration in the pharyngeal phase.
The patient's height and body weight were 85.0 cm and 11.6 kg, respectively. Her weight-for-height Z-score (−0.3) was in the 15th–50th percentile of the World Health Organization (WHO) growth standards [5]. Nutritional evaluation showed that the patient needed a regular daily intake of 989 kcal. However, she could only consume 700 kcal/day, which was less than the required caloric intake. The patient was given supplementary balanced nutrition twice a day.
The patient received an intensive dysphagia rehabilitation program for 30 minutes a day, five times a week, for 6 weeks, and it comprised both direct and indirect interventions, such as oromotor facilitation, thermo-tactile stimulation, and lip strengthening, and compensatory techniques, such as postural modification and alteration of the feeding utensil to facilitate swallowing [6]. She was given anti-spasticity drugs for managing spasticity and achieving a stable posture during feeding. After the treatment, she was fed in a semi-reclined position, which reduced the signs of aspiration and the amount of food or liquid leakage from the mouth.
However, her nutritional status showed no improvement, with her daily calorie intake decreasing to 600 kcal/day after 4 weeks of admission. At 6 weeks postadmission, her total intake further decreased to 392 kcal/day. Her body weight continuously declined from 11.6 kg to 10.5 kg, and her weight-for-height Z-score (−1.6) decreased to the 3rd–15th percentile of the WHO growth standards, which was a sharp decrease of one major percentile line in a short period [5].
Gastrostomy was therefore recommended, but her parents refused the procedure. She was then referred to another hospital for treatment of deconditioning due to dehydration, and she received gastrostomy.
DISCUSSION
The most common initial symptoms in infantile Sandhoff disease include spasticity, increased myotatic reflexes and excessive startle response [7]. In our case, the patient initially presented with spasticity and equinus foot deformity, without an excessive startle response. Firstly, clinicians need to rule out the possibility of hereditary spastic paraplegia with lower extremity hyperreflexia, and spasticity through SPG3A and SPG4 sequencing. Furthermore, clinicians should rule out the possibility of cerebral palsy by clinical regression in the developmental area.
Bley et al. [8] reported that an exaggerated startle response is observed at an average age of approximately 7.2 months and the average onset of spasticity occurs at 13.4 months in the infantile type of Sandhoff disease. Patients showed an average onset of the symptom of aspiration at 22 months of age, with 75% of them undergoing gastrostomy. Our patient also had severe swallowing difficulty at 22 months of age.
The most commonly reported cause of death is primary disease, followed by aspiration pneumonia [8], which means many patients with Sandhoff disease have swallowing difficulty. Therefore, conservative management along with prevention of fatal complications such as aspiration pneumonia is very important. But there are few reports about swallowing difficulty along with its clinical manifestations and management.
There are no established treatment strategies for Sandhoff disease. Various treatment regimens, such as hexosaminidase infusion, leukocyte transfusion, and bone marrow transplantation, have been attempted [9], but none of these treatment regimens have shown successful outcomes. Therefore, patients with Sandhoff disease are given genetic counseling and other supportive management. Oral sensorimotor intervention is the treatment approach in children with oropharyngeal dysphagia in a contemporary clinical setting, but there is not enough high-quality evidence of the effectiveness of this intervention. Children with dysphagia are often unable to consume sufficient energy and nutrients from an oral diet and they require supplemental non-oral feeding, such as nasogastric tube and supplementary parenteral nutrition or in severe cases, a gastrostomy [6].
Our patient presented with dysphagia, which may be due to problems in the oral phase of swallowing rather than in the pharyngeal phase of swallowing. Although her parents wanted to continue oral feeding, several factors such as progressive disease, caregiver burden, duration of feeding, and quality of life were considered, and a gastrostomy, being the optimal feeding technique, was recommended.
In our case, the patient had a prompt worsening of nutritional status as well as dysphagia; it highlights the importance of a regular follow-up and a prompt intervention along with an active assessment of dysphagia and the nutritional status. To the best of our knowledge, this is the first Korean report of an infantile case of Sandhoff disease presenting with dysphagia and change in the nutritional status.
Notes
CONFLICT OF INTEREST: No potential conflict of interest relevant to this article was reported.