INTRODUCTION
Global developmental delay (GDD) and intellectual disability (ID) are major neurodevelopmental disorders in children. About 25%-50% of moderate to profound GDD/ID result from genetic etiology [1]. Among the possible aberrations in chromosomes, submicroscopic rearrangements including subtelomeric microdeletions or microduplications have been identified in GDD/IDD with detection rates ranging from 3% to 23% [2].
The 5q14.3 microdeletion syndrome has recently been recognized as a clinical syndrome, manifesting with severe intellectual disability, febrile seizures, muscular hypotonia, and minor brain anomalies [3]. The myocyte enhancer factor 2C gene (MEF2C), located in chromosome 5q14.3, is considered to be the gene mainly responsible for 5q14.3 microdeletion syndrome [4].
We present a patient with typical features of the 5q14.3 microdeletion syndrome, which has not been reported in Korea yet.
CASE REPORT
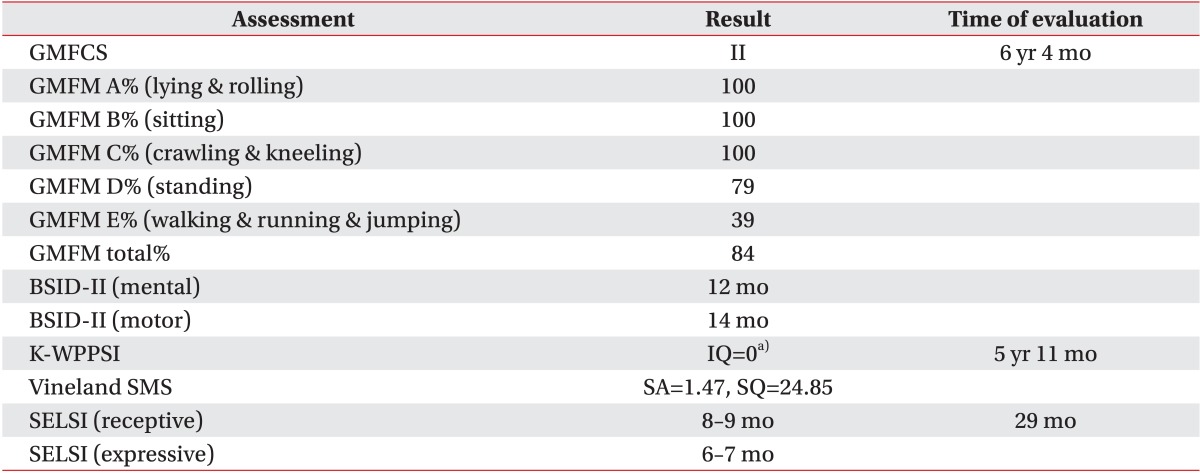
A 6 years 7 months old female patient was born at full term following an uneventful pregnancy as the only child from healthy non-consanguineous parents. Her birth weight was 2,470g (3rd centile), length was 46 cm (3rd centile), and occipital-frontal circumference was 32.5 cm (10-25th centile). She achieved head control in an erect position at 5 months and side-rolling at 7 months, showing a mild developmental delay. Despite having received physical and occupational therapy since the age of 8 months, she showed a GDD when we thoroughly reevaluated her developmental status at 6 years 4 months. All the domains including gross motor, fine motor, cognition, speech, and social behavior were severely delayed (Table 1). Climbing up the stairs alone or standing on one leg was impracticable. She also had problems in planning a series of movements. Fundamental sensory integration of vestibular, proprioceptive, and tactile stimuli were disrupted, leading to poor hand-eye coordination. She paid little attention to any linguistic stimuli, including calling her name, and could not recognize her family. Although there were some vocalizations, there was no meaningful expressive language observed, despite normal audiologic assessment findings. She was not toilet-trained and continued to use diapers. She had trunk hypotonia with mild cervicothoracic scoliosis (Fig. 1A), bilateral coxa valga (Fig. 1B), and pes planus (Fig. 1C). She had 6 febrile convulsions with partial seizures confirmed in an electroencephalography (EEG) at 5 years of age, but a followup EEG one year later showed no epileptiform activity. Both visual and brainstem auditory evoked potentials taken at 22 months were normal. Brain magnetic resonance imaging revealed delayed myelination at both 10 and 19 months (Fig. 2A, 2B), but no focal abnormality at 48 months (Fig. 2C). On ophthalmologic investigation, bilateral esotropia was observed, which required an early corrective surgery at 8 months. She had no prominent dysmorphism in her face except a broad forehead and mildly depressed nasal bridge.
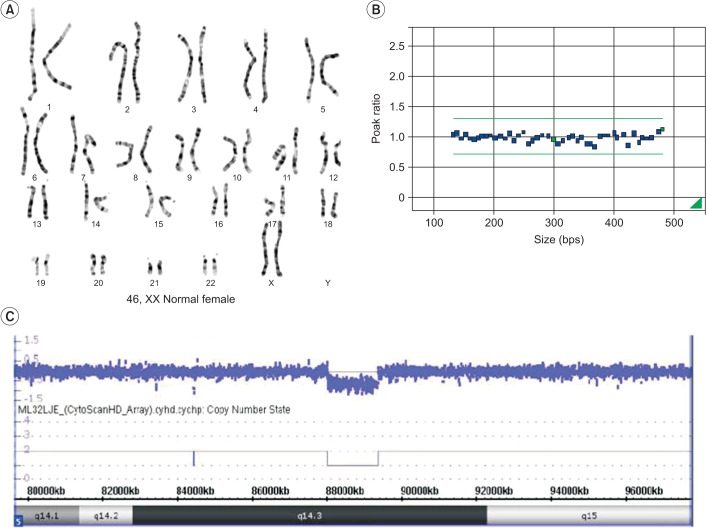
Both chromosomal analysis of the proband and multiplex ligation-dependent probe amplification (MLPA) analysis using a subtelomeric probe set (P070) showed an apparently normal female karyotype (Fig. 3A, 3B). However, on an assessment with array comparative genomic hybridization (CGH), a microdeletion in the chromosomal 5q14.3 region (Fig. 3C) was detected. Both of her parents showed a normal hybridization pattern in that region (data were not shown). The deleted region was about 1332.682 kb in size, starting from 88031637 and ending at 89364319, and according to the ISCA database, it included only one known OMIM gene, MEF2C. The number of probes was 928. The proband's karyotype was 46,XX.arr 5q14.3(88031637-89364319)x1 dn.
DISCUSSION
A 1.33-Mb sized microdeletion in chromosome 5q14.3 region of our patient can be interpreted to be de novo, because such an aberration was not detected in the array CGH of her parents.
MEF2C, located in 5q14.3, consists of 11 coding exons spreading along approximately 100 kb of genomic DNA [4]. MEF2C encodes a transcription factor, which belongs to the myocyte enhancer factor 2 subfamily, participating in the transmission of extracellular signals to the genome and in the activation of the genetic programs that control cell differentiation, proliferation, morphogenesis, survival, and apoptosis of a wide range of cell types [5]. Due to MEF2C's involvement in many organs during various developmental stages, functional disruption of MEF2C results in a wide scope of detrimental effects throughout the course of development.
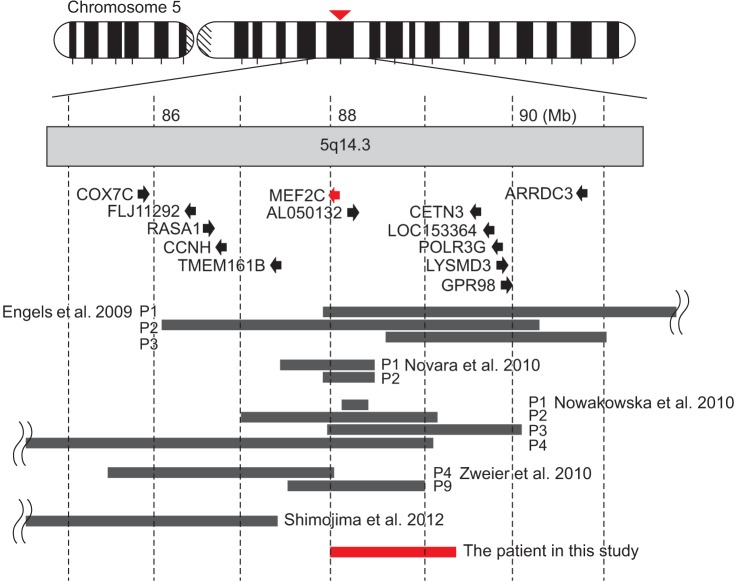
There have been 23 patients with 5q14.3 deletions previously reported worldwide at present [6]. The clinical variability of these patients may be partly influenced by differences in the deletion's size [7]. However, despite having similar deletions in size, the severity of brain anomalies varies considerably from case to case. For example, Novara et al. [8] reported a patient (Fig. 4, P2 of Novara et al.) who had only a small deletion of 318 kbp, but showed severe perinatal hemorrhage and subsequent leukoencephalopathy.
Nowakowska et al. [7] reported that inclusion of MEF2C in the deleted segment is the most essential factor determining clinical features. In cases where MEF2C is not involved, the chromosomal breakpoints were near the MEF2C. Shimojima et al. [6] insisted that the relative location of the deleted segment, that is, whether upstream or downstream of MEF2C, is important in the resulting clinical features and severity. Zweier et al. [4] suggested that genomic regions 233-kb to 500-kb upstream of MEF2C may be required for proper MEF2C expression. According to the Saitsu et al. [9], intellectual disability and early-onset epilepsy are closely related to a disruption of the upstream region of MEF2C.
Compared to previously reported cases, our patient had a relatively small-sized deletion, containing only one gene, MEF2C (Fig. 4). The upstream region from MEF2C was also included in the deleted segment. This result corresponds with previous research which considered MEF2C to be the solely responsible gene for all the symptoms of 5q14.3 deletion syndrome.
In this case, we were able to detect the genetic abnormality, not by subtelomeric MLPA or high resolution chromosome analysis, but only by array CGH. Array CGH has been widely used as a promising diagnostic method for patients with unexplained GDD/ID [10]. Array CGH can detect copy number variations (CNVs) as small as 1 kb, which were recently considered to be the etiology of neurodevelopmental disorders including ID, GDD, autism, and schizophrenia [10]. Array CGH reliably identifies all known microdeletion and microduplication syndromes with additional information, including the exact location, size, and involved genes of the lesion, which may not be detected by conventional cytogenetic methods at all [11]. Thus, some researchers suggest array CGH should be the first-tier test prior to MLPA or G-band karyotyping in order to reduce the total cost of genetic testing, when patient does not present with features of a recognizable genetic syndrome or specific tests for a suspected disorder have been negative [11].
In conclusion, we reported the first patient in Korea with a 5q14.3 microdeletion in which MEF2C resided. By the aid of array CGH, we found the exact location and the size of chromosomal abnormality to establish the cause of the unexplained GDD/ID in this patient. This not only enabled us to make an early diagnosis and set up treatment strategies, but helped to obtain a deeper understanding of the role of MEF2C.