INTRODUCTION
Rett syndrome is a progressive neurodevelopmental disorder in females and a common cause of mental retardation.1 The prevalence is one per 10,000 female children, and mutation of the MECP2 gene in the X chromosome is known to be involved.2 Children with Rett syndrome usually undergo normal development in the early stages of growth but subsequently regress in areas of motor and language. Appropriate hand usage becomes difficult due to stereotypic hand movements as retardation of head circumference growth ensues. Other clinical features such as loss of communication ability, ataxia, autistic tendencies and epilepsy have been reported to occur.1,3 Children with Rett syndrome typically progress through four stages; the early stagnation stage (6-18 months), the second regression stage (1-4 years), the third pseudostationary stage, and the fourth late motor deterioration stage.4 Based on these clinical features, international congresses have reported on revised clinical diagnostic criteria for Rett syndrome.5 However, there is ambiguity in interpreting the criteria. Stagnation of head circumference is not a feature that is observed in all Rett syndrome children and developmental delay can occur early after birth. In addition problems in gait development may be due to ataxia.6 These variations and ambiguities make diagnostic decisions difficult for clinicians when examining children with suspected Rett syndrome.
Most Korean studies on Rett syndrome have reported on the genetics of Rett syndrome or have been studies of electroencephalographic results. None have focused on the clinical features of Korean Rett syndrome patients. The aim of this study is to confirm the known clinical features of Rett syndrome patients who visited our rehabilitation clinic and further focus on functional outcomes as well as contribute to decision making in the clinical field.
MATERIALS AND METHODS
We recruited 26 patients with mutations of the MECP2 gene from 2001 to December of 2010, who visited the Rehabilitation Department of National Health Insurance Corporation Ilsan Hospital and who were diagnosed due to clinical features with classic Rett syndrome.7,8 All patients were followed up to a stage where they would be past the regression stage, which was at least 5 years and older. They were all female children, with ages averaging 60.4 months and ranging from 18 to 176 months at their first visit to our hospital.
The birth history and medical history were investigated with additional histories on development, regression and onset of regression. We surveyed periods where each developmental areas had peaked and also looked for presence of developmental regression, as well as regression periods through caregiver interview, questionnaires, and longitudinal review of medical records on growth parameters and physical examination. Based on normal developmental curves, we classified the patients by their weights and heights as groups of less than 3‰, 3-10‰, 10-25‰, 25-50‰, 50-75‰, and 75-90‰. We defined patients as growth retarded if the patients were either consistently included in the less than 3‰ group or developmental delay group. Hand function regression was used to describe those patients who had lost their ability to reach or grasp. Regression of language was used to define loss of vocalization of formerly able words. Patients who had lost their ability to stand, walk, and change position independently were defined to have regression in gross motor activities. The patients were evaluated through careful history taking and follow up by a rehabilitation specialist in our hospital's outpatient clinic. We also investigated the prevalence of characteristic clinical features of Rett syndrome such as sterotype, growth retardation in head circumference, epilepsy, and scoliosis. Furthermore, we analysed associations between functional levels (independent walking, independent standing, independent sitting, unable to sit) and specific clinical features.
SPSS PC 16.0 was used for the statistical analysis, and descriptive statistics were applied for analysis of growth parameters, prevalences of specific clinical features and analysis of functional levels. Chi-square test was used to analyze correlations between final functional levels and degree of scoliosis and t-test was applied to find correlations between final functional level and onset of epilepsy. The significance level was set at p<0.05.
RESULTS
There were no risk factors in birth history related to neuro-developmental disease such as preterm birth, low birth weight, perinatal asphyxia, and neonatal convulsion. However, there was one patient with a history of neonatal jaundice treated by phototherapy. There were no significant findings related to family or gravidity.
During follow up, 13 patients (50.0%) showed growth retardation in height, and 15 patients (57.7%) in weight. 12 patients (46.2%) showed microcephaly of less than 3‰, but due to the lack of head circumference data at birth, changes in head circumference could not be analyzed. Among the 9 patients with initial normal head circumferences, 4 patients had circumference regression to less than 3‰.
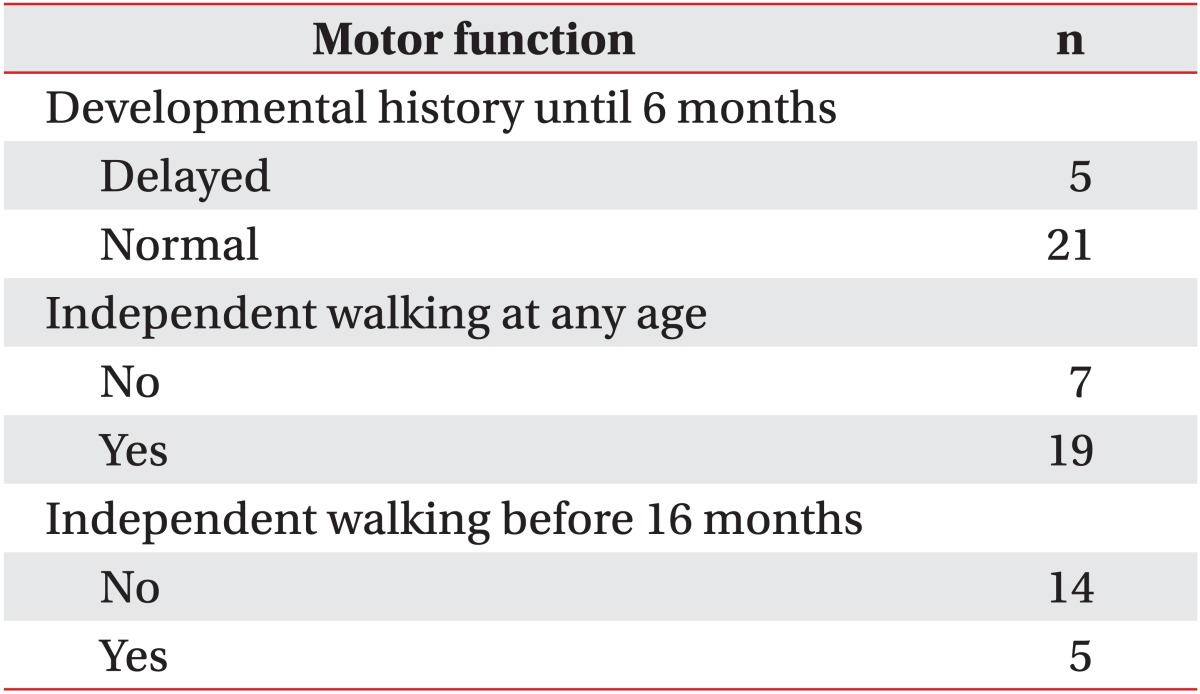
When gathering developmental history, most caregivers were unable to remember periods of eye contact, cooing etc. However, most were able to consistently remember milestones related to gross motor development. Gross motor development during the initial 6 months after birth were normal in 21 patients (80.7%) and delayed in 5 patients (19.2%). Independent walking had been possible in 19 patients (73.1%) where as 7 patients (26.9%) had never experienced independent walking. Only 5 patients (26.3%) could walk normally before 16 months, and the other 14 patients (73.7%) could walk after 16 months (Table 2).
Developmental regression was seen in all the patients, and the average onset age of the regression was 49 months. Among regressive features, regression in hand function was most common and was seen in 21 patients (80.8%). Regression in language development was seen in 16 patients (61.5%) and gross motor development in 8 patients (30.8%).
20 (76.9%) patients manifested with epilepsy that required antiepileptic medication. The onset age of epilepsy was 61-72 months in 4 patients, within 12 months in 2 patients, and 73-84 months and more in 4 patients (Fig. 1).
16 patients (61.5%) had scoliosis with a Cobb's angle of 10 degrees or more at the age of 5, and the angle got larger progressively by serial follow up.
Final functional status of the patients was as follows. Indoor independent gait was possible in 14 but was not in 9 patients. Among the 9 patients who were unable to independently walk, independent standing was possible in 4 patients and independent sitting in 5 patients. 3 patients were unable to sit independently. Final functional status was used to divide the patients into high and low function groups depending upon independent walking status. Developmental histories and clinical features between each group were compared.
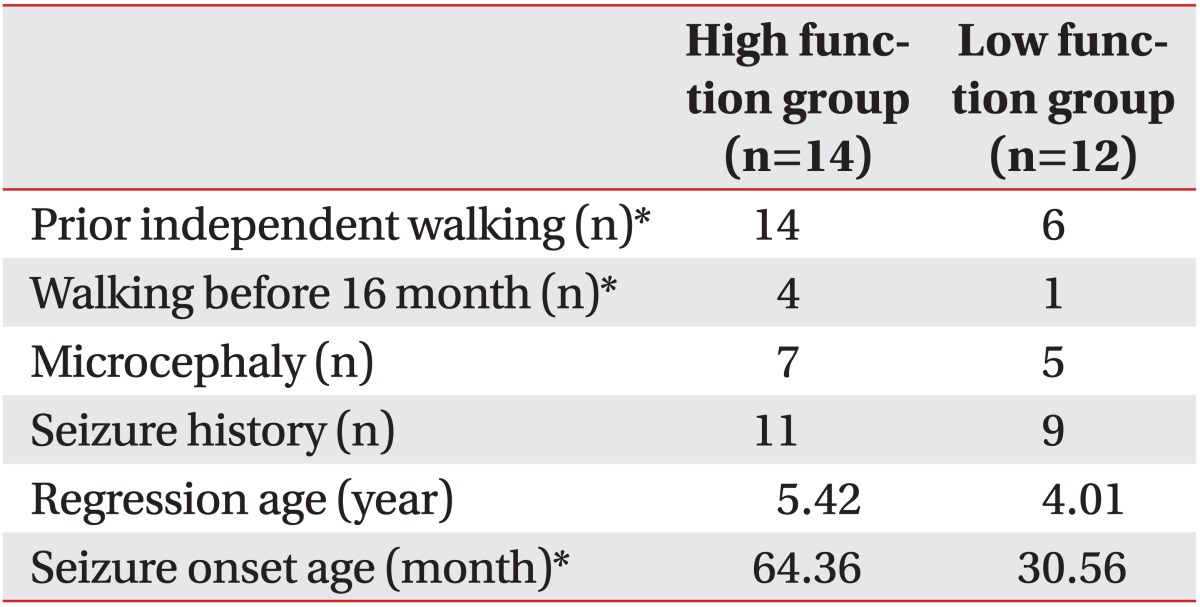
In the high function group, microcephaly with head circumferences of less than 3‰ was observed in 7 (50.5%) out of the 14 patients. Five (41.7%) out of the 12 patients had microcephaly in the low function group. There was no statistical difference between the two groups (Table 3). The high function group included 4 patients (28.6%) capable of independent walking before 16 months and 10 patients (71.4%) capable of independent walking after 16 months. In the low function group, there were 1 (8.3%) and 5 (41.7%) patients who could walk independently before and after 16 months respectively. Six (50.5%) patients had never experienced walking in the low function group. When comparing independent walking status before and after 16 months, there was statistical difference between the low and high function groups (p<0.01) (Table 3). In addition, there was statistical difference in the developmental status of independent walking between the high and low function groups (p<0.01) (Table 3).
The onset of the regression in hand function, language, or gross motor activities was 5.4 years in the high function group and 4.0 years in the low function group, showing a later onset trend in the high function group. However, these differences had no statistical significance (Table 3).
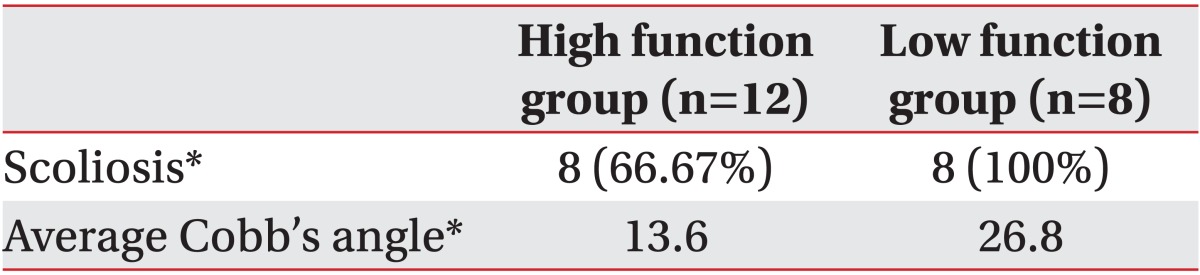
We also analyzed the development of scoliosis according to functional levels and prevalence. Cobb's angle was higher in the low function group but there was no significant difference between the two groups (p<0.05) (Table 4).
There were 11 patients (78.6%) and 9 patients (75.0%) with histories of epilepsy in the high and low function group respectively but prevalences did not show statistical difference. The onset of epilepsy was 64.4 months in the high function group and 30.6 months in the low function group, showing statistically significant differences between the two groups (p<0.05) (Table 3).
DISCUSSION
Rett syndrome was first reported by Andrea Rett in 1960s and gained interest after Hagberg et al.9 reported on characteristic clinical features of Rett syndrome in 1983. The mutation of the MECP2 gene located on Xq28 was revealed to be the cause of Rett syndrome in 1999, but mutation of the MECP2 gene is only an assistive clue in the diagnosis of Rett syndrome since diagnosis is based on clinical criteria.2,3 According to the modified diagnostic criteria presented by Hagberg in 2002, these are the necessary conditions for diagnosing classic Rett syndrome; 1) normal prenatal and perinatal history, 2) normal development during the first 6 months, 3) normal head circumference at birth, 4) growth retardation in head circumference after birth, 5) loss of voluntary hand function during age 6 months through 2 years, 6) stereotypical movement of hands, 7) problems in communication or social interaction, regression of language, and cognitive problem, and 8) regression or impairment in movement. Elsewhere, there are 8 assistive and 5 exclusive conditions. In this study, we recruited cases that corresponded with classic Rett syndrome and cases with mutation of the MECP2 gene. Clinical features were examined since the criteria described above have areas of ambiguity in clinical application. There were some cases where growth retardation in head circumference, which is known to be a necessary condition for the diagnosis, was not definite. In addition there were cases where abnormal development occurred in the early days after birth, and cases where independent walking was impossible due to apraxia, etc.
In this study, about half of the patients showed microcephaly, and among the patients with known head circumferences at birth, half were proven to have subsequent microcephaly. These results correspond to previous studies indicating that although growth retardation of head circumference is a necessary condition for the diagnosis of Rett syndrome, it is not observed in all patients.10 The revised diagnostic criteria for Rett syndrome by the Rett Search Consortium in 2010 excluded growth retardation of head circumference after birth as an essential criterion for the diagnosis of Rett syndrome.11
Patients with classic Rett syndrome have no specific problems in the course of birth and are known to undergo normal development for 6 months after birth. However, some recent studies report on tendencies of decreased interaction with the environment, autistic features, and decreased muscle tone. Moreover, one study revealed that the patients' general movements were abnormal during the first 6 months via video analysis.12 Our study was highly dependent on caregiver reports on the early developmental periods of the patients. Although efforts were made to retrieve information on various developmental milestones, there were limitations and only major gross motor developmental status was analyzed. As a result, according to caregiver descriptions, most of the patients underwent normal development in early periods after birth, and only 19% were reported to have delayed development. Other studies showed similar results with normal psychiatric development seen in 70% and a 2 to 4 month delay in gross motor development seen in 30%.13 Although it is difficult to conclude definitely on early period gross motor delay in children with Rett syndrome, many studies are reporting abnormal development in the early periods of Rett syndrome.
In this study, 26.9% of patients had never walked independently. This result corresponds to results by Smeets et al.14 reporting that 29% of Rett syndrome patients had been unable to walk independently.
In this study, the average age of regression onset was 49 months with regressions most frequently occurring in areas of hand function. Downs also reported that 40% of Rett syndrome patients showed loss of hand function at the age of 3 to 4.15 According to a long term study in Sweden, regression begins at 19 months on average, and at the age of 2.5 years, apraxia or involuntary stereotype is observed in all patients.16 This study shows an earlier age of regression onset compared to our study. According to Segawa, seterotypical hand movements in Rett syndrome patients may be due to a dysfunction in the supplementary motor area which provides antagonistic signals to the hand opposite of that performing voluntary movement. Such dysfunctions of the supplementary motor area and premotor cortex can occur due to problems in the dopamine pathway of the basal ganglia or substantia nigra.17 Among the subjects included in this study, regression symptoms were recognized earlier in those diagnosed with Rett syndrome at our hospital's rehabilitation clinic, whereas regression periods in those diagnosed at other departments and institutions were detected at a later stage. This study has limitations in that it relied primarily on caregiver descriptions in the absence of video interpretations or other objective data. This may be due to a lack of education about Rett syndrome among the general population, delaying the detection and downgrading the seriousness of atypical behaviors of children.
Epilepsy is known to develop in 60-80% patients of Rett syndrome, and this study showed similar percentages.4,18 However, the onset age of epilepsy was somewhat different. It is known that the prevalence of epilepsy increases after the third regression period and in a study by Glaze et al.,18 epilepsy rarely develops before the age of 2 years. However in our study, epilepsy occurred evenly in all ages. Although epilepsy is known to be related to the patients' functional level, we found no differences in the prevalence of epilepsy between the high and low function group. Rather, only onset age of epilepsy differed in these two groups, with epilepsy occuring later in high function group than in the low function group. Although there are studies reporting on the absence of an association between onset age of epilepsy and clinical severity, a number of studies are showing an increased incidence of epilepsy in Rett syndrome as age increases.
The most common musculoskeletal complication in Rett syndrome is scoliosis, and the prevalence varies according to reports but ranges from 48-87%.19 The prevalence of scoliosis increases as developmental disability severity increases. It also increases when regression occurs before 6 months, when there has been no prior experience of walking, or when regression of gait has begun before musculoskeletal maturation.20 These findings, according to Ager and Alan, are due to mutations of R294X and R306C in the MECP2 gene in chromosome Xq28, which are related to mild developmental problems and to a decreased risk of scoliosis development.21,22 In this study, about 60% of patients had scoliosis, and it was more frequent in the high function than in the low function group as in other studies. Riise et al.23 also reported that in Rett syndrome patients where scoliosis was not a problem, all were capable of independent walking. Thus rehabilitative strategies are needed to prevent and treat scoliosis in Rett syndrome patients who are unable to walk.
CONCLUSION
Through this study, we were able to identify characteristic features of Rett syndrome as well as confirm that ambiguous descriptions included in the widely known diagnostic criteria of Rett syndrome are not always observed in all patients. Taken together, the 2010 revised diagnostic criteria of Rett syndrome is more appropriate when diagnosing Rett syndrome.
Furthermore, decrements in functional decline after the age of 5 were more profound in the low function group in whom independent gait was not possible. In addition, the onset of epilepsy was earlier, and scoliosis more severe.
This study can be applied in the rehabilitation field to minimize disability in patients with Rett syndrome.